The advanced therapy medicinal products (ATMPs) represent a fast-growing field with a lot of potential in severe, rare, or chronic diseases for which no adequate conventional treatments exist. The ATMPs can be classified into three main types: gene therapy products, cell-based products, and tissue-engineered products. In addition, some ATMPs may contain one or more medical devices as an integral part of the product, which are referred to as combined ATMPs. An example of that is the cells embedded in a biodegradable matrix or scaffold.
Given the heterogeneity and the complexity of the ATMPs, the conventional strategies designed for robust non-clinical (NC) assessment of proof-of-concept (PoC), mechanism of action (MoA), toxicology and bio-distribution are not always directly transferable into the ATMP development. The standard non-clinical in vitro, and in vivo testing of the dose-related safety and efficacy have had limited value for the ATMP, specifically, due to the different immunologic backgrounds and microenvironments for this class of compounds.
One of the differences in the development strategy for the ATMPs compared to small molecule or biologics is the fact that there are few specific guidelines describing the type of studies that should be performed. Therefore, the ATMPs NC studies are dependent on a thorough risk assessment of the effects of the product. As a result, the design of the safety and toxicity studies are different for the ATMPs than for other types of compounds, e.g., tissue selection for the pharmacokinetics studies, animal model assessment, potency assays validation, proof-of-concept studies, etc. There can be significant uncertainties with the translation of the study data from the NC studies to the human situation, which might influence the ATMP benefit-risk assessment. Therefore, a major challenge is to identify platforms enabling rigorous evaluation of the NC outcomes, which are meaningful and predictive for human clinical trials. Animal models are still quite common as part of the NC studies, although alternative in vitro assays become more and more available as substitutes for the animal use.
Recently, the USA has adopted legislation opening the way to perform non-human safety studies without performance of studies in animals before starting clinical studies or marketing approval. There are many initiatives to replace animal testing in Europe as well. How quickly regulators will be fully confident in relying on safety data only captured via in vitro or in silico data is not yet clear. However, the use of new “human-similar” methods, such as human mini-organs ("organoids"), organ-on-a-chip and computer-based methods instead of the standard animal tests will most probably have a large impact on the speed of the development and associated costs.
The RBA is the dominant strategy of the ATMPs development, and is used to determine the extent of quality, non-clinical and clinical data to be included in the Marketing Authorization Application dossier. It includes the identification of various risks associated with the clinical use of the specific ATMP and risk factors inherent to the ATMP with respect to quality, safety, and efficacy. ‘Risk’ is defined as a “potential unfavorable effect that can be attributed to the clinical use of ATMP and is of concern to the patient and/or to other populations, include e.g., unwanted immunogenicity, disease transmission, tumor formation.
The exact package of non-clinical safety studies to be performed (studies with animals and/or alternative models) will be dependent on the outcome of the risk analysis. The number of studies to be performed will probably be smaller than for small molecules and not extra compared to biologicals. There are specific aspects to be covered as well e.g., biodistribution and shedding.
To overcome those hurdles, the ATMP developers need to foster collaborations with industry partners and engage with regulatory agencies to define, evaluate, and develop appropriate NC models where relevant data is unavailable.
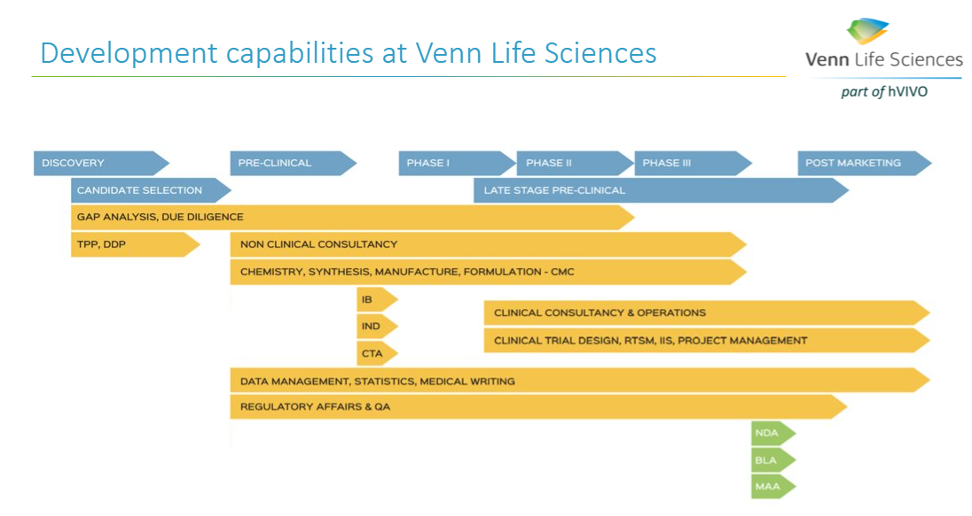
A successful ATMP development requires a solid plan (e.g., Drug Development Plan (DDP) based on the RBA), that outlines each step for the advancing of a new product from the lab to the envisioned marketed ATMP. The DDP is a living document and should include the roadmap for the activities to be performed (all pharmaceutical, non-clinical, clinical studies and regulatory activities), the description of the ATMP development strategy to a certain milestone (FiH, PoC, IND/CTA, MAA/NDA), the inventory of work, and associated timelines and costs based on the risk assessment. The DDP, together with a thorough RBA, can help in convincing (potential) investors on the thoroughness of the project and would thereby facilitate the funding of the project.
Identifying all those steps early is an essential part of a successful ATMP development. At Venn Life Sciences, the team of experienced consultants have the expertise to help with the various steps in the ATMP development and marketing approval, including helping to define the intended target population, and the desired product attributes (indication or usage & dosage, administration & pharmacology, adverse reactions & toxicology, safety, and efficacy).