Selecting a Contract Development and Manufacturing Organization (CDMO) can be challenging, especially given today’s evolving geopolitical landscape. These seven tips can guide you toward a successful CDMO partnership.
1) Cost-effectiveness
Conducting clinical trials in Asia often proves significantly more cost-effective when partnering with a local CDMO, largely due to lower production costs. Asian CDMOs that have undergone FDA inspections are typically viewed as more reliable – while pricier than other in this region, they still offer savings compared to their counterparts in the US and EU.
2) Quality
Having robust quality systems in place is crucial. EU and US CDMOs are often more trusted than their Asian counterparts due to regular regulatory inspections. Their established standards offer clients greater assurance, whereas some Asian CDMOs may still require closer supervision to meet similar expectations.
3) Regulatory alignment
Production materials intended for the EU or US markets must comply with the quality standards set by their respective regulatory authorities. Therefore, regulatory alignment is essential. In such cases, conducting at least part of the clinical development in the EU or US might provide strategic advantages.
4) Reliability
Drug product materials must meet high standards, and the supply chain should be reliable. Lower-cost production – such as with certain Asian CDMOs – necessitate additional oversight and control mechanisms. Conducting a quality assurance (QA) audit early in the selection process is highly recommended to ensure compliance and reliability.
5) Risk management
Implementing process controls and risk management early in the manufacturing process allows for the identification of critical parameters at an early stage, thereby streamlining the upscaling process.
6) Standardisation
Use of common techniques and appropriate equipment in the production process will support a seamless transfer to future larger-scale commercial manufacturing.
7) Timeliness of delivery
Adhering to strict timelines for on-time delivery and avoiding any material shortages during a clinical trial will ensure smooth progression of clinical trials and overall project success.
By thoughtfully evaluating these tips, the selection process of a CDMO will become more manageable and successful. Partnering with experienced experts and adhering best practices are crucial for overcoming challenges and ensuring a cost-effective and well-organized supply chain.
If you're evaluating CDMO partners, we can help you make the right choice; guided by strategy, experience, and global insight.
Contact one of our experts today!
Advanced Therapy Medicinal Products (ATMPs) are human medicines based on cells, genes, or tissues and are seen as pioneering new opportunities for the treatment of a range of diseases. Currently, 22 ATMPs have been approved by the FDA, and the field of interest is growing rapidly. In traditional drug development, pharmacokinetic (PK) studies are necessary to use medicines under the best conditions of efficacy and safety.
For ATMPs, investigations of PK are also essential aspects as delineated in EMA’s guide on ATMPs, including flowcharts and checklists. However, there could be significant differences compared to traditional ADME (Absorption, Distribution, Metabolism, and Excretion) studies, and special considerations unique to the respective ATMPs are required. This is because ATMPs may not follow the common principles of ADME or exhibit a classical dose-response relationship. For example, in the case of cell therapies, clinical PK studies may consider body distribution/migration, cells growth, viability, differentiation/proliferation, and/or phenotype, depending on what is relevant to the specific ATMP.
Among 22 ATMPs approved by the FDA, five are chimeric antigen receptors (CAR) T-cell therapies: tisagenlecleucel (Kymriah™), lisocabtagene maraleucel (Breyanzi®), axicabtagene ciloleucel (Yescarta ™), idecabtagene vicleucel (Abecma®), and brexucabtagene autoleucel (Tecartus™). In this blog, we shed light on CAR-T therapy as an example case for ATMP drug development.
CAR-T therapy involves genetically engineering and expanding autologous (or, in the future, potentially allogeneic) T-cells, after which the modified T-cells are administered to the patient. The genetically engineered CAR-T cells are adapted to target tumour cells using the CAR, which binds to specific antigens on tumour cells. After infusion, the concentration of CAR-T cells observed in the blood declines as the CAR-T cells distribute into the tissues. Upon interaction with the tumour target, the CAR-T cells become activated and rapidly expand, which can be measured as a rise to maximum concentrations in the blood (1-2 weeks after infusion). After this peak, the concentrations slowly decline, reflecting the death of the CAR-T cells (e.g. due to a decrease in tumour burden). However, CAR-T cell concentrations may continue to be observed for years and can even increase again when available target tumour cells increase. Since the engineered CAR gene is directly related to the expansion and persistence of CAR-T cells, PK studies are necessary in the development of these ATMPs.
Therefore, unlike most ATMP products, the PK of CAR-T cells involves the evaluation of traditional ADME PK parameters, but these need to be interpreted in the context of cellular kinetic parameters indicative of expansion (Cmax) and persistence (AUC, tlast). At Venn Life Sciences, our team of experienced consultants and data analysts has the expertise to support and manage PK analyses for new therapies like ATMPs.
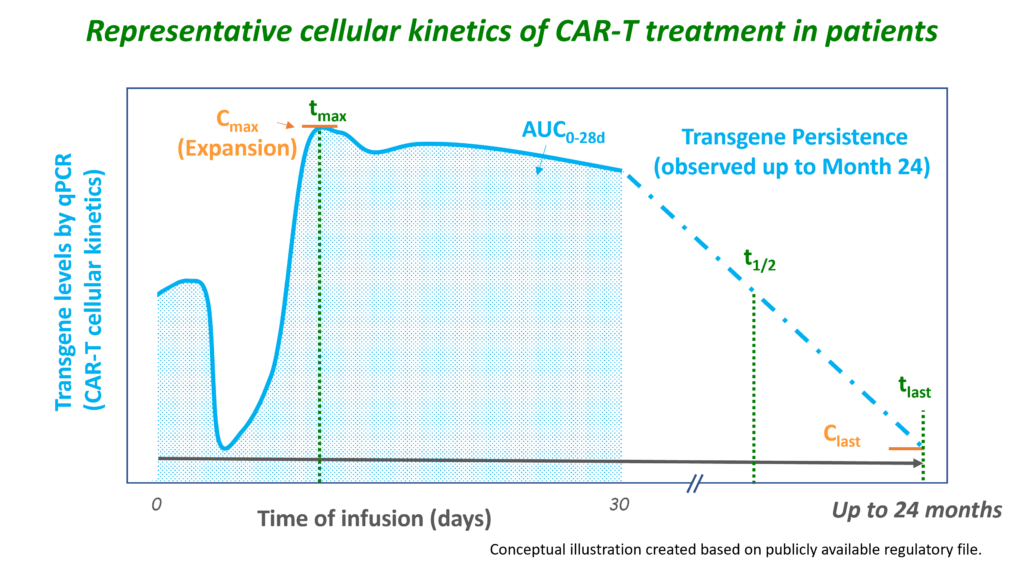
The PK profiles of CAR-T cells do not always reflect a traditional PK profile, and therefore the following practical aspects need to be taken into account:
If you are interested in learning more about how we can support you with PK in ATMPs, please contact Venn Life Sciences at BD@vennlifesciences.com. We would welcome the opportunity to discuss PK in ATMPs and other related aspects.
The advanced therapy medicinal products (ATMPs) represent a fast-growing field with a lot of potential in severe, rare, or chronic diseases for which no adequate conventional treatments exist. The ATMPs can be classified into three main types: gene therapy products, cell-based products, and tissue-engineered products. In addition, some ATMPs may contain one or more medical devices as an integral part of the product, which are referred to as combined ATMPs. An example of that is the cells embedded in a biodegradable matrix or scaffold.
Given the heterogeneity and the complexity of the ATMPs, the conventional strategies designed for robust non-clinical (NC) assessment of proof-of-concept (PoC), mechanism of action (MoA), toxicology and bio-distribution are not always directly transferable into the ATMP development. The standard non-clinical in vitro, and in vivo testing of the dose-related safety and efficacy have had limited value for the ATMP, specifically, due to the different immunologic backgrounds and microenvironments for this class of compounds.
One of the differences in the development strategy for the ATMPs compared to small molecule or biologics is the fact that there are few specific guidelines describing the type of studies that should be performed. Therefore, the ATMPs NC studies are dependent on a thorough risk assessment of the effects of the product. As a result, the design of the safety and toxicity studies are different for the ATMPs than for other types of compounds, e.g., tissue selection for the pharmacokinetics studies, animal model assessment, potency assays validation, proof-of-concept studies, etc. There can be significant uncertainties with the translation of the study data from the NC studies to the human situation, which might influence the ATMP benefit-risk assessment. Therefore, a major challenge is to identify platforms enabling rigorous evaluation of the NC outcomes, which are meaningful and predictive for human clinical trials. Animal models are still quite common as part of the NC studies, although alternative in vitro assays become more and more available as substitutes for the animal use.
Recently, the USA has adopted legislation opening the way to perform non-human safety studies without performance of studies in animals before starting clinical studies or marketing approval. There are many initiatives to replace animal testing in Europe as well. How quickly regulators will be fully confident in relying on safety data only captured via in vitro or in silico data is not yet clear. However, the use of new “human-similar” methods, such as human mini-organs ("organoids"), organ-on-a-chip and computer-based methods instead of the standard animal tests will most probably have a large impact on the speed of the development and associated costs.
The RBA is the dominant strategy of the ATMPs development, and is used to determine the extent of quality, non-clinical and clinical data to be included in the Marketing Authorization Application dossier. It includes the identification of various risks associated with the clinical use of the specific ATMP and risk factors inherent to the ATMP with respect to quality, safety, and efficacy. ‘Risk’ is defined as a “potential unfavorable effect that can be attributed to the clinical use of ATMP and is of concern to the patient and/or to other populations, include e.g., unwanted immunogenicity, disease transmission, tumor formation.
The exact package of non-clinical safety studies to be performed (studies with animals and/or alternative models) will be dependent on the outcome of the risk analysis. The number of studies to be performed will probably be smaller than for small molecules and not extra compared to biologicals. There are specific aspects to be covered as well e.g., biodistribution and shedding.
To overcome those hurdles, the ATMP developers need to foster collaborations with industry partners and engage with regulatory agencies to define, evaluate, and develop appropriate NC models where relevant data is unavailable.
A successful ATMP development requires a solid plan (e.g., Drug Development Plan (DDP) based on the RBA), that outlines each step for the advancing of a new product from the lab to the envisioned marketed ATMP. The DDP is a living document and should include the roadmap for the activities to be performed (all pharmaceutical, non-clinical, clinical studies and regulatory activities), the description of the ATMP development strategy to a certain milestone (FiH, PoC, IND/CTA, MAA/NDA), the inventory of work, and associated timelines and costs based on the risk assessment. The DDP, together with a thorough RBA, can help in convincing (potential) investors on the thoroughness of the project and would thereby facilitate the funding of the project.
Identifying all those steps early is an essential part of a successful ATMP development. At Venn Life Sciences, the team of experienced consultants have the expertise to help with the various steps in the ATMP development and marketing approval, including helping to define the intended target population, and the desired product attributes (indication or usage & dosage, administration & pharmacology, adverse reactions & toxicology, safety, and efficacy).
The clinical development of Advanced Therapy Medicinal Products (ATMPs) can be a daunting task, but partnering with a team of experts can pave the way for success. In this blog, we highlight ten key considerations that can make the development process less risky and more efficient.
ATMP clinical trials often use donor or patient-based starting materials, leading to inherent variability. Defining release specifications for cell-based ATMPs must take this variability into account to ensure consistency and efficacy.
Due to the scarcity of ATMP materials, retaining samples for reconfirmation may pose challenges. Developing a sampling strategy that aligns with product stability and shelf-life is essential to maintain quality assurance.
ATMP development involves bidirectional traceability and technical demands for coding, processing, preservation, storage, and distribution of human tissues and cells. Complying with these requirements ensures product safety and transparency.
The process of taking biopsies or extracting cells may carry risks for the subject and impact the product's quality and safety. Careful consideration and mitigation strategies are necessary to safeguard both patients and product integrity.
Detailed procedures for ATMP handling, disposal, preparation, and bedside administration are critical. Additionally, healthcare professionals should receive proper training to ensure safe and effective administration.
Determining the appropriate ATMP dose and repeatability can be complex. Thorough research and scientific expertise are vital to achieve optimal treatment outcomes.
Previous or concomitant treatments and prior infections or vaccinations can significantly impact ATMP efficacy. A thorough assessment of these factors is essential for accurate clinical trial results.
Considering potential long-lasting and/or irreversible effects on clinical trial subjects, their close contacts, and future pregnancies is crucial. Comprehensive risk analysis should be performed to ensure patient safety.
Planning for long-term safety follow-ups after ATMP treatment is essential. This includes reporting and monitoring serious adverse reactions and events, with investigators well-trained in adverse event reporting.
A differentiated causality assessment is necessary for each component of the ATMP, such as the cell-based and medical device components in combined ATMPs. Additionally, product administration procedures, concomitant medication, and product failure must be thoroughly evaluated.
By carefully considering these ten key aspects, the clinical development of ATMPs can become more manageable and successful. Collaboration with experienced experts and adherence to best practices will play a vital role in overcoming challenges and ensuring the safe and efficient advancement of ATMPs.
Don't miss our exclusive webinar where we delve into the intricacies of ATMPs development and manufacturing on 19 September 2023.
Sign up now
As a medicinal expert at one of the leading notified bodies of the EU, I’ve been involved in the assessment of medicinal files of many different device-drug combination products. These are devices that include ancillary medicinal substances, in which the device has the primary mode of action, and the medicinal substance has an additional beneficial effect. The ancillary medicinal substances range from small molecules to human blood derivatives and recombinant products. Depending on the nature of the ancillary medicinal substance a consultation with a national competent authority or the EMA is required. Human blood derivatives and recombinant products are higher risk products and therefore require assessment by EMA. The consultation process for medical devices is comparable to filing a market application.
The notified body acts as liaison between the manufacturer and the competent authority, the consultation process is conducted by the notified body and therefore the aim of the notified body review is to ensure that the dossier complies with the requirements of the competent authority. These requirements are similar to the requirements of dossiers for medicinal substances (CTD format) and additionally, the notified body includes a usefulness report. This usefulness report gives a summary on the risks and benefits of the inclusion of the ancillary medicinal substance and will only be generated if the benefit is considered to outweigh the risk.
When there are changes to the device that affect the ancillary medicinal substance a supplementary consultation (variation) may be required. The classification of the change on the ancillary medicinal substance is assessed using the EMA classification guideline for human medicines. Based on the type of change and the competent authority that is used for the initial consultation a judgement is made if a supplementary consultation (variation) is required. In some cases, a change would be reviewed in-house and the outcome of the review would be shared with the competent authority to check if they agree with the assessment and agree that this change can be approved without a supplementary consultation.
Some drugs use devices, such as syringes or nasal sprays for example, as administration tools. Since the implementation of the medical device regulation in May 2021 a notified body opinion (NBOp) is required in the market application dossier for devices that are integral to drugs. This is an assessment which provides a statement on the conformity of the device part with the relevant GSPRs as set out in Annex I of the MDR. As a medicinal expert, I was involved in reviewing the article 117 applications from a medicinal perspective to identify the risks/hazards associated with administration of the drug that needed to be addressed in the documentation of the device. In summary, I have extensive experience in interacting with the major EU competent authorities and EMA. I know what is expected to be included in the medicinal dossiers for medical devices that include ancillary medicinal substances. And additionally, I have experience in performing reviews for drug-device combinations that fall under article 117 of the MDR, and therefore will be able to advise what needs to be included in the technical file for the device of the drug-device combination.

There are several distinctive features that must be considered in the ATMP development:
Have you experienced additional difficulties in your ATMP development? We would love to learn more about your development program and explore how we can impact your work.
Let us facilitate your ATMP advancement in the minimum of time and with the most efficient use of resources, while ensuring the high quality. It would be great to establish a cooperation between Venn Life Sciences and your company.
Discover how we can support and accelerate the ATMP development:
Ion Tcacencu (MD, PhD) is an ATMP expert with over 17 years of experience as scientist within regenerative medicine and immunology, being responsible for projects developing new ATMPs.

Appropriate scale-up/down is key for further larger production. Important is to focus to the right parameters that can help us to set the design for all experiments at lab scale.
The common approach within the up-stream processing (USP) part in biologics is to increase the scale 10 times. It means from e.g. 25L lab scale to max. 250L pilot scale and later on from pilot scale to max 2500L production scale. Key parameters are vessel geometry, agitation, aeration, back-pressure and feeding profiles (in case of fed-batch fermentation). Next diagram shows typical construction of a common fermenter with key parameters:
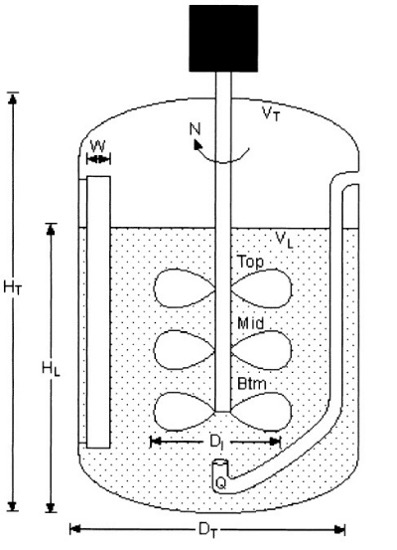
Geometric similarity of fermenter geometry is a pre-requisite for applying established scale-up relationship. It´s expressed as follows: DT2/DT1 = (VT2/VT1)1/3, where DTi is fermenter diameter and VTi total fermenter volume. Geometric similarity also assumes reasonably constant impeller geometry such as impeller diameter (DI) and number of impellers (N). Based on target total a/o working volumes obtained from geometric similarity, the desired working volume in the fermenter may be altered during experimentation. The ratio of the impeller to fermenter diameter (DI/DT) in standard fermenters is between 0.3 to 0.45. Fermenters with a standard geometry are beneficial within scale-up correlations assuming constant geometry. Common pilot scale-up fermenters have HT/DT ratios of 3:1, but they can also decrease to 1:1.
The first approach within agitation is to check the design of impellers, number of impellers, diameter and location. Common impellers used within microbial fermentations are Rushton turbines or Hollow-blade (U-shape), but could also be Hydrofoil, Maxflo, etc. Stirrer tip speed (STS) is the simplest approach normally used in case of same design of impellers between two scales. It is formulated as STS = πNIDI, where π is a constant, NI and DI are the impeller speed (s-1) and impeller diameter (m) in fermenter respectively. Typical STS ranges from 3.8 to 7.6 m/s.
More complex approach within agitation is Constant (gassed) power input per liquid volume (PG/VL) which characterizes energy generated by impeller to liquid volume used in the fermenter. It is normally used in a case of scale-up for various design of impellers between two scales. PG/VL is expressed as PG/VL = P0/VL*0.5 = ((NPNI3DI5ρ)/VL)*0.5, where NP is the power number, which means proportionality factor based on impeller design (NP for Rushton is 5 and for Hollow-blade is 1.5). NI and DI are the impeller speed (s-1) and impeller diameter (m), ρ is specific broth density (kg/m3) and VL is volume of fermentation broth (L). General values of PG/VL in large scale (fermenter with a total volume more than 1500L) are between 1 to 3 W/L. It´s difficult to have high power per unit volume at the large scale due to practical limitation of the motor size.
Additional scale-up parameters are focused to oxygen transfer. They are Oxygen Uptake Rate (OUR) and Mass transfer coefficient (KLa). Scale-up based on OUR assumes that the OUR is equal to Oxygen transfer rate (OTR). This equation is expressed as OTR = KLa(csat – cL) = OUR = μX/YX/O2 , where csat is the broth dissolved oxygen (DO) at saturation, cL is the measured broth DO concentration, μ is the specific growth rate, X is the measured cell density and YX/O2 is the cell yield calculated per amount of consumed oxygen. There are several correlations for the determination of KLa using Gassed power per liquid volume (PG/VL) described previously and Superficial gas velocity VS, where the formula looks is KLa = f2(PG/VL)aVSb , where f2 is a proportionality constant. In general, the “a” value within this equation decreases with increasing working volume. The “a” value is 0.95, 0.67 and 0.5 for fermenters from lab scale (e.g. 10L scale), pilot scale (300L) and production scale (more than 20,000L) respectively. Same holds for the “b” value: 0.67 for lab and pilot scale and 0.50 for production scale. The formula for Superficial gas velocity (VS) is VS = φG/(π/4)*DI2), where φG is Gass flow rate (m3/s) and DI is impeller diameter (m). Finally Gass flow rate can be calculated using φG = (Q * (tact/t0)*(Patm/Pabsolute))/3600, where Q is airflow rate (Nm3/s), tact and t0 are actual temperature and temperature of absolute ZERO in Kelvin and finally Patm and Pabsolute are the pressures. Production fermenters up to 100,000L scale have KLa value between 400 to 800 h-1. Scale-up based on KLa is complicated approach by the fact that it is process specific and it changes over the fermentation, making it difficult to reliably quantify. Alternatively, measurement of broth DO concentration (CL) can be used as an adequate scale-up parameter. The minimum acceptance value of CL is well known from lab scale experiments. Cascade control of DO by agitation, aeration and back-pressure can be effective in maintaining of DO above critical value.
The Table 1 shows an example of most important parameters within scale-down model from 300L pilot fermenter to 75L lab fermenter. Appropriate set of all required parameters in small scale can be predictive for consecutive production in large scale. Finally, all set process parameters need to be confirmed in larger scale via pilot or ENG/Validation batches within further cGMP production.
CMC experts from Venn Life Sciences have strong expertise within theoretical and practical application of these models. Do you need more insight or support with your up-scaling process? Do not hesitate to contact us via our website: www.vennlifesciences.com.
Table 1. Practical application of scale-down model from pilot fermenter (300L) to laboratory fermenter (50L).
| 300L pilot fermenter | Fermenter parameters used for Scale down | 75L lab. fermenter |
| 3.32 | Ratio HT/DT | 3:1 |
| 200L | Working volume Vmax | 50L |
| Rushton, 3 times | Impeller type and No. | Rushton, 3 times |
| 0.23 | Impeller diameter DI | 0.12 |
| 4 kW | Engine power PE | 1.1 kW |
| 22 kW/m3 | Engine input per working volume PE/VL = PE/(Vmax/1000) | 20 kW/m3 |
| 420 rpm = 7 rps | Agitation Nmax | 800 rpm = 13.33 rps |
| 5.06 | Stirrer tip speed STSmax = πNmaxDI | 5.03 |
| 16.8 Nm3/h = 280 L/min | Airflow rate Qmax | 6 Nm3/h = 100 L/min |
| 1.40 vvm (1/min) | Volumetric airflow rate per volume Qmax/Vmax | 2 vvm (1/min) Scale down recalculation to 1.4 vvm corresponds to airflow 70 L/min |
| 5.15 W/L (for Nmax) | Constant (gassed) power input per liquid volume P0/VL*0.5 = ((NPNI3DI5ρ)/VL)*0.5 | 5.50 W/L (for Nmax) Scale-down recalculation to 5.15 W/L corresponds to agitation 782 rpm |
Note: For successful scale down model is necessary to set appropriate parameters in lab. scale such as agitation, aeration and back-pressure, that mimic parameters in pilot scale.
Date: 24.10.2022
Author: Juraj Boylo
Gene therapy offers significant potential to treat diseases with high unmet medical need. However, the unique nature of these therapies poses challenges in product development, namely: safety concerns, efficacy issues, or obstacles related to Chemistry, Manufacturing and Controls (CMC). CMC is an integral aspect of gene therapy development. Done right, CMC development of complex manufacturing processes and analytical testing panels can accelerate clinical development and bring these important medicines to patients faster. For gene therapy products, CMC development often needs to be done in a highly compressed timeline, and there are important CMC challenges that need to be addressed to bring these products into the clinic and beyond, amongst which:

Gene therapy products are similar to other biological products in many ways. CMC hurdles in development of biologicals/ biotechnological products are common to our Venn experts; they have key experience in providing support in the development of a wide range of complex biological products. Their broad expertise allows them to have a holistic view on the CMC development of gene therapy products, and to provide out-of-the-box solutions. In example, their experience with human adenovirus-based vaccines allows to translate this expertise to adeno associated virus (AAV)- based gene therapies. The Venn CMC team is well aware of the requirements for early (Phase I) and later (Phase II and III) phases of development and can support you in CMC challenges you are facing in your gene therapy development. The Venn CMC team can help the client to find the optimal solutions to drive your product in development to clinical success.
One of the first steps is creating a solid roadmap for your product, a drug development plan, which outlines your drug development strategy and contains a carefully planned set of steps and action plans, progressing from non-clinical to the first-in-human Phase I to Phase II “proof of concept” and pivotal Phase III trials for registration. Our Venn experts can guide through your journey and can built a comprehensive drug development strategy for your product.
The next step is to support you in early and timely discussions with regulatory agencies on your development plan. Priority for agencies is to keep patients safe. Regulators want to see that a sponsor has control over its products and processes in development. Sponsors should therefore take advantage of, e.g., Innovation Task Force meetings and scientific advice meetings with the EMA or EU national authorities, and/ or INTERACT and Pre IND meetings with the US FDA, to seek advice and alignment with the agencies on CMC challenges you are facing, and the solutions and path forward you are proposing. The Venn regulatory team can plan and manage your health authority interactions. Our consultants are experienced in generating scientific advice/ Pre-IND briefing packages and liaising with US and EU regulatory agencies. They can also act as sponsor representative during EU scientific advice procedures.
Once you have tackled the CMC hurdles and you have buy-in from the agencies on your drug development plan, you can start planning the first-in-human Phase I trial for which you need to produce GMP-compliant investigational product, and for which you need to submit a clinical trial application. The Investigational Medicinal Product Dossier (IMPD) in the EU, or Investigational New Drug (IND) dossier in the US, is one of several pieces of the investigational product related data required for this application. The IMPD/ IND includes information related to the quality, manufacture, and control of the investigational product (including placebo). Guidance on the structure and CMC data requirements for investigational gene therapy products exists for EU and US applications. The EU and US guidelines for investigational
gene therapies have been built on the existing IMPD/ IND guidelines for biologicals/ biotechnological products, and the Venn CMC team is well equipped to support you in authoring the IMPD/ IND.
In addition to the clinical trial application, gene therapy products classified as genetically modified organisms will be subject to a GMO application to the applicable biosafety agencies in the EU to permit the release of the product into the environment (so-called biosafety review). Pending on the modalities of the use, the product can be considered to be contained use or deliberate release, each coming with a different set of requirements. The expectations for environmental risk assessments and GMO applications are regulated on a national level and the requirements and classification can therefore vary significantly between member states in the EU, resulting in a maze of requirements that are difficult to navigate. The Venn regulatory team can guide you in this process and can help you with the procedural aspects of the GMO application and clinical trial application.
In conclusion, there are important CMC challenges that need to be addressed to bring a gene therapy product into the clinic and beyond, but many aspects of gene therapy development are similar to other biological products. With guidance and expertise of our Venn experts in the biological/ biotechnological field, your chances of a successful outcome of your gene therapy development will be increased.

